About Malonylation
 Protein post-translational modifications (PTMs) play an essential part in the regulation of cellular functions and are associated with many disease processes [1-4]. Malonylation is a newly discovered lysine PTMs identified by mass spectrometry and protein sequence-database searching [5]. It is confirmed that malonylation is related to many biological processes. For example, malonylation was demonstrated to be a potential role in type 2 diabetes as a result of the malonylated proteins enrichment in metabolic pathways, especially the pathways involved in glucose and fatty acid metabolism [6]. Reversible protein malonylation also regulates other key cellular pathways including the urea cycle and other mitochondrial enzymes [7]. Furthermore, lysine malonylation was reported to be a new type of histone PTMs, and that aberrant modification of histones contributes to diseases, including cancer [8].
Protein post-translational modifications (PTMs) play an essential part in the regulation of cellular functions and are associated with many disease processes [1-4]. Malonylation is a newly discovered lysine PTMs identified by mass spectrometry and protein sequence-database searching [5]. It is confirmed that malonylation is related to many biological processes. For example, malonylation was demonstrated to be a potential role in type 2 diabetes as a result of the malonylated proteins enrichment in metabolic pathways, especially the pathways involved in glucose and fatty acid metabolism [6]. Reversible protein malonylation also regulates other key cellular pathways including the urea cycle and other mitochondrial enzymes [7]. Furthermore, lysine malonylation was reported to be a new type of histone PTMs, and that aberrant modification of histones contributes to diseases, including cancer [8]. Asides from the studies have been performed in bacteria, mouse, and human, the biological functions of malonylation in plant are similarly attention. For instance, malonylation is proved to be a key reaction in the metabolism of xenobiotic phenolic glucosides in Arabidopsis and tobacco [9]. Additionally, by the experiments on lysine malonylaome in Triticum aestivum L. and Oryza sativa, analyses showed predominant presence of malonylated proteins in metabolic processes, including carbon metabolism, glycolysis/gluconeogenesis, TCA cycle, as well as photosynthesis[10, 11].
[1]Nørregaard Jensen, O., Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Current Opinion in Chemical Biology, 2004. 8(1): p. 33-41.
[2]Wang, Y.-C., S.E. Peterson, and J.F. Loring, Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Research, 2013. 24: p. 143.
[3]Ahearn, I.M., et al., Regulating the regulator: post-translational modification of RAS. Nature Reviews Molecular Cell Biology, 2011. 13: p. 39.
[4]Gong, C.X., et al., Post-translational modifications of tau protein in Alzheimer’s disease. Journal of Neural Transmission, 2005. 112(6): p. 813-838.
[5]Peng, C., et al., The first identification of lysine malonylation substrates and its regulatory enzyme. 2011. 10(12): p. M111. 012658.
[6]Du, Y., et al., Lysine malonylation is elevated in type 2 diabetic mouse models and enriched in metabolic associated proteins. Mol Cell Proteomics, 2015. 14(1): p. 227-36.
[7]Nishida, Y., et al., SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol Cell, 2015. 59(2): p. 321-32.
[8]Xie, Z., et al., Lysine Succinylation and Lysine Malonylation in Histones. Molecular & Cellular Proteomics, 2012. 11(5): p. 100-107.
The flowchart of Kmalo
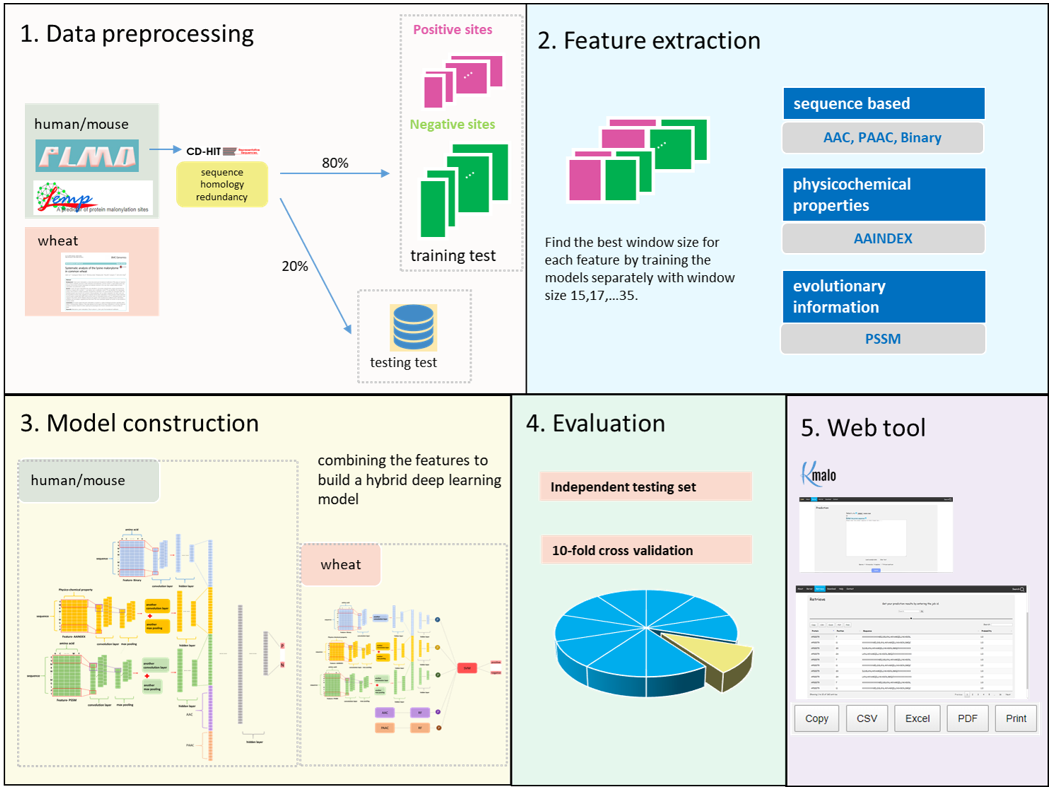
Data Resource

Homo sapiens

Mus musculus

Triticum aestivum
For mammalian data, we collected the dataset in PLMD and LEMP. PLMD is an online data resource specifically designed for collecting protein lysine modifications from different public databases and literature, which includes 5013 validated malonylation sites from 1841 H. sapiens proteins and 4390 malonylation sites from 1466 M. musculus proteins. LEMP is the latest web tool for predicting malonylation sites in mammalian proteins. It used 5288 malonylation sites and 88636 non-malonylation sites for training and testing the prediction model and freely provided the data for research purposes on its website.
For plant data, we collected plant proteins from a recent study (Liu, J., Wang, G., Lin, Q. et al. BMC Genomics (2018)) which identified 342 malonylation sites in 233 T. aestivum proteins with a systematic analysis.
Model construction
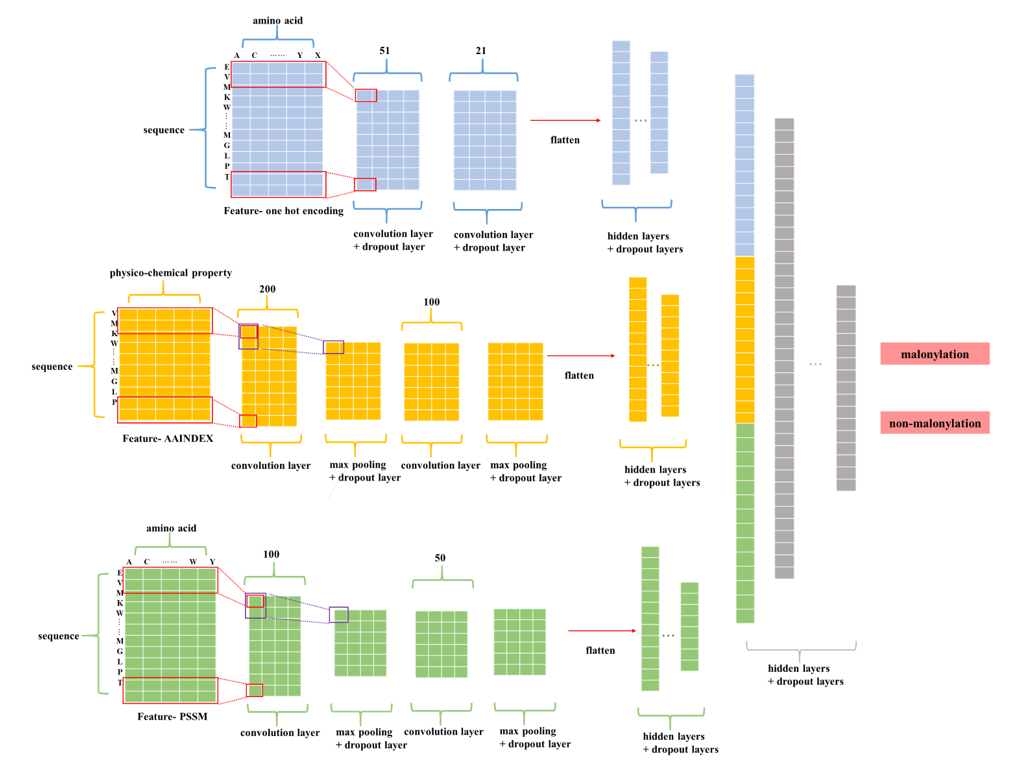
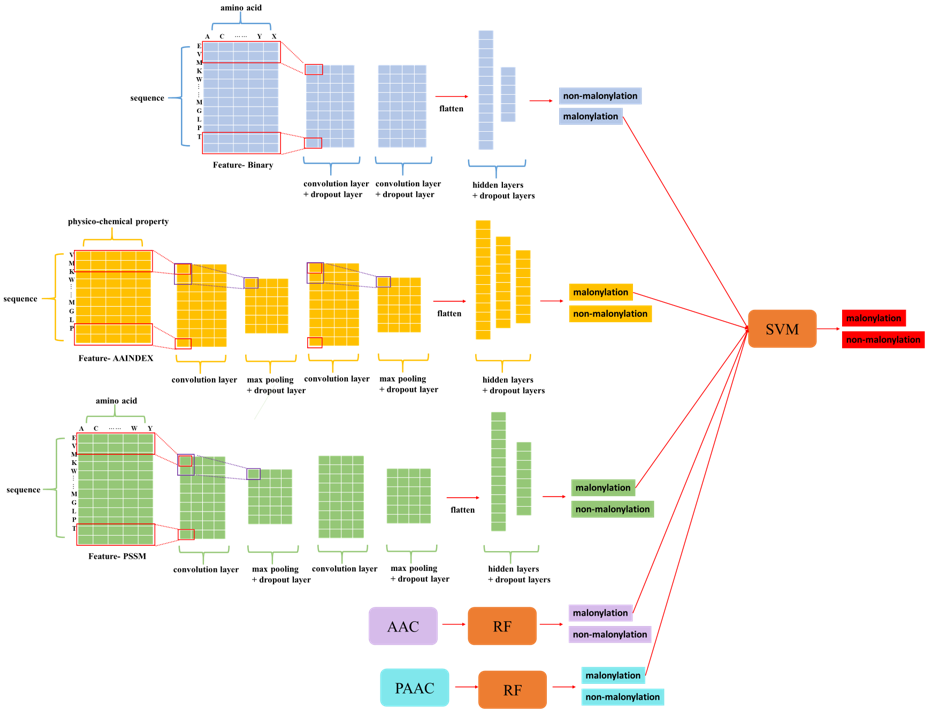
The purpose of the second part was to aggregate the separated models trained by different features to make a final predicted decision. There were three methodologies for aggregation: another neural network, SVM, and majority votes. For the mammalian group, we found out that seeing the CNN models as a feature extractor and combing together to feed into another deep learning neural network returned the best AUC. We also noticed that using feature one hot encoding, AAindex, and PSSM profile three features has a better AUC than using all five features. As for the plant group, feeding the positive probability of each feature to a SVM model reached the best performance. The cutoff threshold is set to 0.046 for mammalian model, 0.195 for plant model.